Along with classical QTL mapping in biparental populations—where we phenotype and genotype every individual—there has always been a smart shortcut: Bulk Segregant Analysis (BSA).
Instead of analyzing all individuals, BSA pools DNA from individuals with extreme phenotypes (e.g., highly resistant vs highly susceptible) and compares allele frequencies between these groups.
👉 And today, thanks to the dramatic reduction in sequencing costs, we no longer genotype these pools with a handful of markers—we sequence them.
This is how QTL-seq was born.
🔬 Why QTL-seq matters today
The integration of BSA with next-generation sequencing (NGS) has transformed trait mapping:
✔️High resolution and accuracy
✔️Much faster than traditional QTL mapping
✔️Significantly lower cost
✔️High-throughput and scalable
In fact, these approaches can identify genomic regions controlling traits rapidly and with high precision, often comparable to classical mapping approaches .
For companies, this is key:
👉 Speed + precision + cost-efficiency = competitive advantage
Especially when working with major-effect QTLs like disease resistance genes, and and there is a robust phenotyping protocol that discriminates clearly the extremes.
⚙️ How QTL-seq works (in practice)
The workflow is elegant and efficient:
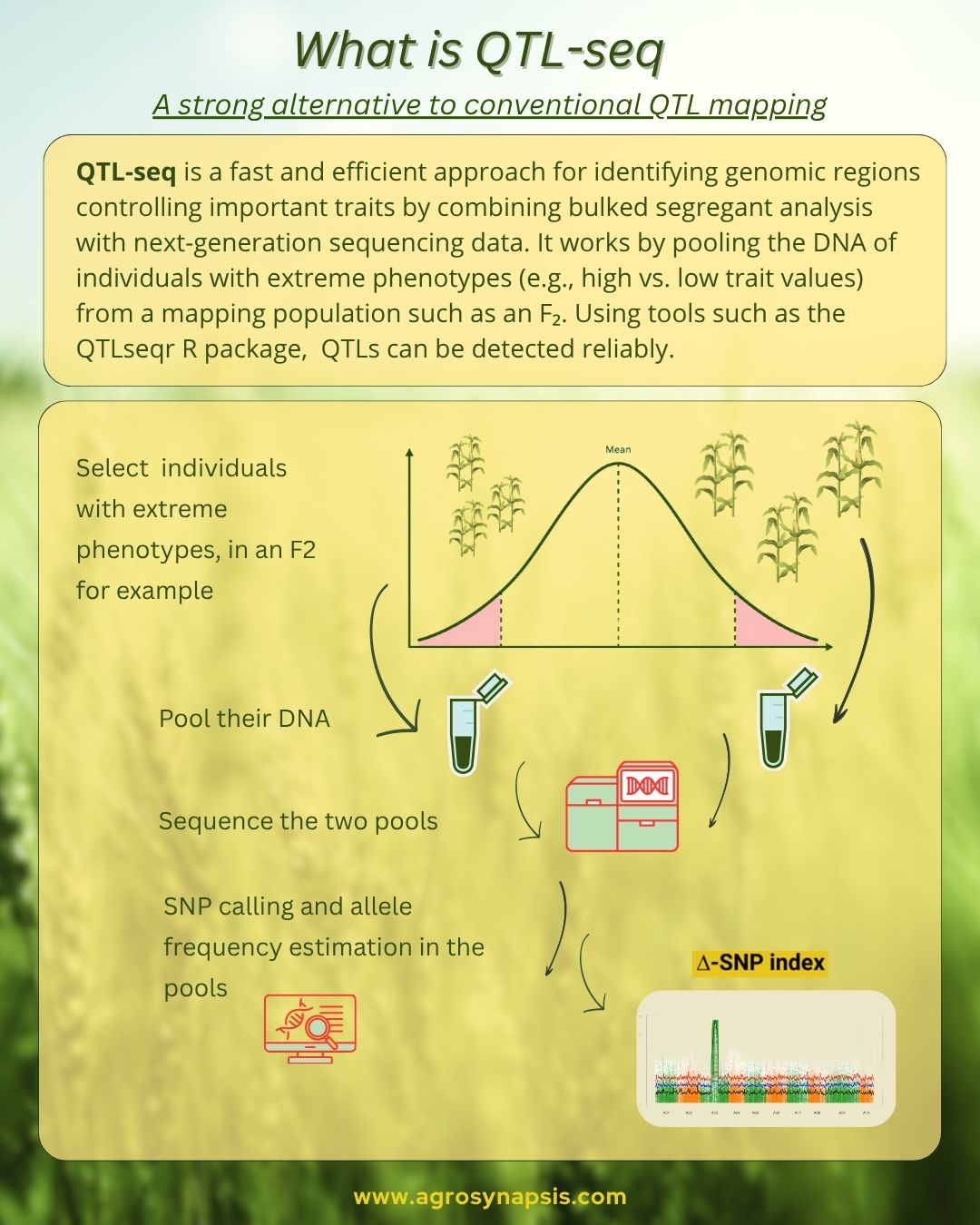
1. Create a segregating population
(e.g., F2 from parents with contrasting phenotypes)
2. Select individuals with extreme phenotypes
(e.g., most resistant vs most susceptible)
3. Pool DNA into two bulks
→ Resistant bulk
→ Susceptible bulk
4. Sequence both bulks
5. Align reads and call SNPs
6. Compare allele frequencies between bulks
→ Calculate SNP-index and ΔSNP-index (for example using the R package QTLseqr)
7. Identify genomic regions (QTLs)
→ Regions with strong allele frequency differences are linked to the trait
The key idea:
Marker alleles associated to the locus controlling the trait become enriched in one bulk, while neutral alleles remain randomly distributed.
📊 Proven in real breeding scenarios
QTL-seq is not theoretical—it’s already widely used:
Disease resistance in groundnut
Fruit traits in tomato
Flowering time and plant height in cereals
Stress tolerance (heat, cold, salinity)
Multiple studies have successfully identified major QTLs and candidate genes across crops using this approach .
📖 For those who want to dive deeper into the methodology and real-case applications, here is an excellent review:
https://lnkd.in/eE-qr3-w
👉 If you’d like to be informed about the upcoming workshops organized by AgroSynapsis, and receive early access and discounts, 𝗳𝗶𝗹𝗹 𝗼𝘂𝘁 𝗼𝘂𝗿 𝘀𝗵𝗼𝗿𝘁 𝘁𝗿𝗮𝗶𝗻𝗶𝗻𝗴 𝗶𝗻𝘁𝗲𝗿𝗲𝘀𝘁 𝗳𝗼𝗿𝗺 here:
https://lnkd.in/g3tApqPz
BLOG ON MOLECULAR BREEDING
QTL-seq: A fast, precise, and cost-effective alternative to biparental QTL mapping for breeding companies
Discover how QTL-seq combines bulk segregant analysis and sequencing to rapidly identify trait-linked genomic regions with high precision and low cost.